Presented by :Dr. S. K. Rai
President Corp. R&D & Technical
Effective Date 31 December 2025
2.
Schedule M, partof the
Drugs and Cosmetics Act of
1940, outlines Good
Manufacturing Practices
(GMP) for pharmaceutical
products in India.
It specifies the requirements
for manufacturing facilities,
equipment, and processes to
ensure consistent quality and
safety of pharmaceutical
products.
3.
Understanding the Timeline of key Events in
Revised Schedule M implementation
The Government of India
notifies the Revised Schedule
M under the Drug and
Cosmetics Act, bringing
India’s Good Manufacturing
Practices (GMP) In line with
global standards.
2023
2024
Large Pharmaceutical
Manufacturers (Turnover
above 250 crore)were
required to comply with
Revised schedule M
standards by this date.
The original compliance
deadline for small and
medium- sized
pharmaceutical
manufacturers (turnover
250 crore or less) was
set for this date.
28 Dec
28 June
4.
The Ministry ofHealth & Family
Welfare announces a deadline
extension for small and medium-
sized pharma manufacturers,
granting them until December 31,
2025, to fully comply with Revised
Schedule M*
There is no blanket extension.
Only if you apply before May 11th,
there is an extension up to year-
end.
Manufacturers must submit
Form A to the central License
Approving Authority
outlining their upgradation
plan. Failure to submit within
this timeframe may lead to
penalties, including potential
factory closures.
The final deadline for all
pharmaceutical manufacturers
(irrespective of turnover)to
achieve full compliance with the
Revised Schedule M.
There is no blanket extension.
Only if you have applied before
May 11th, there is extension up to
year end. If you don’t apply, it
means you have complied.
5.
5
Site Master File
29
SelfInspection and Quality Audit
15
General Requirements
1
Part I A to Part I F – Specific requirements for
manufacture of -
Quality Control System
16
Warehousing Area
2
Sterile Products, Parental Preparations
A
Specifications
17
Production Area
3
Oral Solid dosage forms
B
Master Formula Records
18
Ancillary Area
4
Oral Liquids
C
Packaging Records
19
Quality Control Area
5
Topical Products
D
Batch Packaging Records
20
Personnel
6
Metered Dose Inhalers
E
Batch Processing Records
21
Health, Clothing and Sanitation for
personnel
7
Active Pharmaceutical Ingredients
F
SOPs and Records
22
Manufacturing Operations and Control
8
Part II-
Reference Samples
23
Sanitation in manufacturing premises
9
Requirements for Plant and Equipment
Reprocessing and Recoveries
24
Raw materials
10
Distribution Records
25
Equipment
11
Validation and Process Validation
26
Documentation and Records
12
Product Recall
27
Labels and Printed Materials
13
Complaints and Adverse Reactions
28
Quality Assurance
14
EXISTING SCHEDULE M CONTENTS
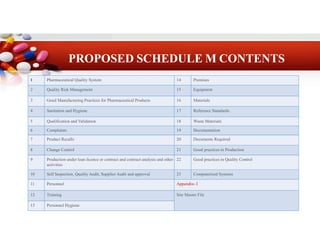
6.
6
Premises
14
Pharmaceutical Quality System
1
Equipment
15
QualityRisk Management
2
Materials
16
Good Manufacturing Practices for Pharmaceutical Products
3
Reference Standards
17
Sanitation and Hygiene
4
Waste Materials
18
Qualification and Validation
5
Documentation
19
Complaints
6
Documents Required
20
Product Recalls
7
Good practices in Production
21
Change Control
8
Good practices in Quality Control
22
Production under loan licence or contract and contract analysis and other
activities
9
Computerised Systems
23
Self Inspection, Quality Audit, Supplier Audit and approval
10
Appendix- I
Personnel
11
Site Master File
Training
12
Personnel Hygiene
13
PROPOSED SCHEDULE M CONTENTS
7.
New Schedule M;Good Manufacturing Practices and Requirements of
Premises, Plant and Equipment for Pharmaceutical Products
7
Part II to Part XII
Specific requirements for manufacture of Sterile Products, Small & Large Volume Parentals, Ophthalmic
Preparations
Part II
Specific requirements for manufacture of Hazardous substances such as Sex Hormones, Steroids or Cytotoxic
substances (Newly Added)
Part III
Specific requirements for manufacture of Biological Products (Newly Added)
Part IV
Specific requirements for manufacture of Radiopharmaceutical Products (Newly Added)
Part V
Specific requirements for manufacture of Phytopharmaceutical Products (Newly Added)
Part VI
Specific requirements for manufacture of Investigational Pharmaceutical Products for Clinical Trials in Human
(Newly Added)
Part VII
Specific requirements for manufacture of Oral Solid Dosage Forms
Part VII
Specific requirements for manufacture of Oral Liquids
Part IX
Specific requirements for manufacture of External Preparations
Part X
Specific requirements for manufacture of Metered Dose-Inhalers
Part XI
Specific requirements for manufacture of Active Pharmaceutical Ingredients
Part XII
8.
New Schedule M;Good Manufacturing Practices and Requirements of
Premises, Plant and Equipment for Pharmaceutical Products
8
Part XIII
Requirements of Plant and Equipment for External Preparations
1
Requirements of Plant and Equipment for Oral Liquid Preparations
2
Requirements of Plant and Equipment for Tablets
3
Requirements of Plant and Equipment for Powders
4
Requirements of Plant and Equipment for Capsules
5
Requirements of Plant and Equipment for Surgical Dressing
6
Requirements of Plant and Equipment for Ophthalmic Preparations
7
Requirements of Plant and Equipment for Pessaries and Suppositories
8
Requirements of Plant and Equipment for Inhalers and Vitrallae
9
Requirements of Plant and Equipment for Repacking of Drugs and Pharmaceutical Chemicals
10
Requirements of Plant and Equipment for Parental Preparations
11
9.
SCHEDULE M- EXISTINGVS REVISED
Revised Schedule M
Existing Schedule M
Revised Schedule M is divided as Part-I to Part XIII
Existing Schedule M is divided in two parts
Part I: Good Manufacturing Practices for Pharmaceutical
Products
Part II to Part XII: Specified requirements for
manufacturing, as per product categories, e.g. Sterile Products,
Oral Sold Dosage Forms etc.
Part 1: Good Manufacturing Practices For Premises and
Materials
Each section is explained with certain requirements but not in
specific details. Subparts are Part IA to IF
Part IF: GMP for API-Specific product Types
Five new categories are added as compared to existing
Schedule M
Part 2: Requirements of plant and Equipment
Part XIII: Requirements of plants and equipment for
manufacturing of 11 categories of pharma products
This section is similar as of previous schedule M 2001.
10.
Concerns and Challengesfaced by MSMEs in implementing the
Revised Schedule M
• Multi product manufacturing facilities and loan licensing
• Batches not produced regularly
• Concept of formulation development
• Excipient compatibilities studies
• Stability studies
• Bioavailability / Bioequivalence studies
• Quality culture at all levels
• Continuous training
• Continual improvement in the quality
• Data integrity
11.
Revised Schedule M:Part-I
PartI:GMP For Pharmaceutical Products:Main Principles
1. Pharmaceutical Quality system (PQS)
2. Quality Risk Management (QRM)
3. Good manufacturing practices for pharmaceutical
products
4. Sanitation and hygiene
5. Qualification and validation
6. Complaints and adverse reaction
7. Product recalls
8. Change control
9. Production under loan license or contract and
contract analysis and other activities
10. Self-inspection, quality audits and suppliers’ audits
and approval
11. Personnel
12. Training
13. Personnel Hygiene
14. Premises
15. Equipment
16. Materials
17. Reference Standards
18. Waste materials
19. Documentation
20. Good practices in production
21. Good practices in quality control
22. Computerized systems & Appendix-
SMF
12.
1.Main Principle’s OfPharmaceutical Quality System (PQS)
Establish Quality manual / document to describe the Organization Quality Management system C all elements like role of
production, quality management responsibility, MRM etc.
Senior management shall involve to effective implementation of PQS.
GMP / GXP should be applied to all stages of product of life cycle.
Product and process knowledge is managed throughout all lifecycle stages.
Product realization is achieved by designing, qualifying, planning, implementation, maintaining and continuously
improving a system.
There is a procedure for self-inspection or quality audit that regularly appraises
the effectiveness and applicability of the product quality system.
Deviations, shall be reported, investigated and recorded appropriate level or root cause and CAPA shall be applied
followed by effectiveness of CAPA shall be monitored.
Periodic management reviews shall be conducted to identify opportunities for continual improvement of products,
processes and PQS.
Quality Risk Management(QRM)-ICH Q9
Product Quality Review shall include:
•Review of critical in-process controls.
• Review of QMS elements, OOS, Deviation,
complaints, returns, recalls, changes etc.
•Review of the result of the stability monitoring
programmed and any adverse trends.
•Review of qualification status of relevant equipment
and utilities, e.g., heating, ventilation and air
conditioning, water or compressed gases and a
review of the results of monitoring the output of
such equipment and utilities.
• Evaluation of the risk to quality is based on
scientific knowledge, experience with the process
considering risk to the patients.
• The level of effort, formality and documentation
of the Quality Risk Management process is
commensurate with the level of risk.
• Regular periodic quality reviews all the
pharmaceutical products, shall be conducted with
the objective of verifying the consistency of the
existing process to identify product and process
improvements.
QRM - Product Quality Review (PQR)
2.Quality Risk Management
All manufacturingprocesses are clearly defined,
systematically reviewed for associated risks in the light
of scientific knowledge and experience, and shown to
be capable of consistently manufacturing
pharmaceutical products of the required quality that
comply with their specifications.
Qualification and validation are performed.
Products are carried out correctly and personnel are
should be trained.
The proper storage and distribution of the products
which minimizes any risk to their quality.
System is available to recall any batch of product from
sale or supply.
3.Good Manufacturing Practices (GMP)
Continued……….
Sanitation coverspersonnel, premises, equipment
apparatus, production materials and containers.
A high level of sanitation and hygiene shall be
practiced in every aspect of the manufacture of
drugs.
The scope of sanitation and hygiene covers
personnel, premises, equipment and apparatus,
production materials and containers, and
disinfection to prevent contamination.
Potential sources of contamination shall be
eliminated through an integrated comprehensive
programmed of sanitation and hygiene.
4.Sanitation and Hygiene
Continued……….
Detailed discussionabout Design qualification (DQ) /
installation qualification (IQ) / operational qualification (OQ) C
Performance qualification (PQ).
Any aspect of operation, including significant changes to the
premises, facilities, equipment or processes, which may affect
the quality of the product, directly or indirectly, shall be
qualified and validated.
The commitment to maintain continued validation status shall
be
stated – Quality manual or validation master plan
5.Qualification andValidation
Continued……….
There shallbe written procedures describing the action to be
taken, including the need to consider a recall, in the case of a
complaint concerning a possible product defect.
The licensing authorities shall be informed if a manufacturer is
considering action following the faulty manufacture, product
deterioration, a suspect product or any other serious quality
problems with a product.
The license shall have a pharmacovigilance system in place for
collecting, processing, and forwarding the reports to the
licensing authorities for information on the adverse drug
reactions emerging from the use of drugs manufactured or
marketed by the license
6.Complaints andAdverse Reactions
24.
Quality complaint: Originateat consumer level and concern with
physical, chemical and biological properties or condition of labeling
and / or packaging of the product.
Adverse Reaction complaints: Due to allergic reactions of any
other untoward reaction or fatal reaction or near fatal reaction.
Other medically related complaint: Include complaints such as lack
of efficacy or clinical response.
Types of Complaints Continued……….
25.
Any substanceintroduced into the body can a risk at normal
doses, and all are potentially toxic if given in overdose.
However, we should keep in mind that the great majority of
treatments are safe and effective if the principles of
pharmacology and pharmacokinetics are carefully applied.
The common risk in terms of side-effects, adverse drug
reactions, and toxicity.
Adverse Reaction Continued……….
26.
The licensingauthorities shall be promptly
informed of any intention to recall the
product because it is , or is suspected of
being defective.
Traceability- Distribution
Communication C Coordination
AOI manufacturer to formulator
Regulatory authorities
Recall committee C action plan
Mock recall
7.Product Recalls
27.
What to becontrolled …………
1.Changes in the raw materials,
2.Specifications,
3.Analytical methods,
4.Facilities,
5.Support systems,
6.Equipment (including computer
hardware),
7.Processing steps,
8.Labelling and packaging materials
9.Computer software.
8.Change Control
28.
Production under loanlicense or contract and contract analysis and other
activities:
GMP is Applicable, .. Describes Contract Production
Contract Analysis
Role and Responsibilities of Contract giver, Contract Accepter
Agreement, Batch release, Technology Transfer, Distribution etc.
9.Contract Production &Testing
29.
Detect anyshortcoming and implementation
of CAPA
Self-inspection team
Frequency of Audit-performed routinely and
in specific occasions i.e. recall or inspection
by licensing authorities. At least once in a
year.
Self-inspection report C Follow-up action
Suppliers audit and approval –RM/PM
Self inspection consists of…..
Periodic detail examination of condition
Working procedure by a team from production site with aim of verifying that good pharmaceutical manufacturing
practice are being applied and purpose any necessary corrective measures to responsible management.
10.Self-inspection & QualityAudit
• The establishsystem of Quality Assurance (QA)
• Necessary qualifications and practical experience.
• Training, including hygiene instruction, relevant to their needs.
• Organization chart, Roles and Responsibilities of QA, QC, Production,
• Key personnel: for supervising the production and QA C QC Shall possess the
qualifications and experience as specified under the rules.
• Define authorized person for approving a batch for release
11.Personnel
34.
Quality Control (QC)in the pharmaceutical industry is essential for ensuring the safety, efficacy, and consistency of
drug products. It involves rigorous testing and monitoring at various stages of production to meet regulatory standards
and prevent the release of substandard or harmful medications.
Role of quality control in pharmaceutical industry
Key Functions of Quality Control in Pharma:
1. Testing and Analysis:
2. Compliance with Regulations:
3. Preventing Defects and Deviations:
4. Ensuring Product Safety and Efficacy:
5. Maintaining Product Quality:
6. Supporting Continuous Improvement:
7. Preventing Recalls and Litigation:
8. Building Trust and Reputation:
9. Raw Material Testing:
10. In-Process Testing:
11. Finished Product Release:
35.
Role of qualitycontrol in pharmaceutical industry
Key Functions of Quality Control in Pharma:
•Testing and Analysis:
QC laboratories conduct physical, chemical, biological, and microbiological tests on raw materials, in-process samples, and finished products to verify identity, purity, potency, and other quality
attributes.
•Compliance with Regulations:
QC ensures adherence to Good Manufacturing Practice (GMP) guidelines and other regulatory requirements set by bodies like the FDA and EMA.
•Preventing Defects and Deviations:
QC aims to detect nd prevent defects or deviations in the manufacturing process through inspections, testing, and analysis.
•Ensuring Product Safety and Efficacy:
QC ensures that medications are safe for patients by preventing contamination, impurities, and other quality issues that could cause adverse effects.
•Maintaining Product Quality:
QC helps maintain consistent product quality by verifying that each batch meets the required standards before release.
•Supporting Continuous Improvement:
QC data and insights can be used to optimize manufacturing processes and improve product quality over time.
•Preventing Recalls and Litigation:
By identifying and addressing quality issues early, QC helps prevent costly product recalls and legal actions.
•Building Trust and Reputation:
Consistent quality control builds trust in pharmaceutical products and enhances the company's reputation.
•Raw Material Testing:
QC verifies the identity, strength, and purity of raw materials before they are used in production.
•In-Process Testing:
QC monitors the quality of the product during various stages of manufacturing.
•Finished Product Release:
QC releases finished products only after they meet all quality standards.
Quality Control (QC) in the pharmaceutical industry is essential for ensuring the safety, efficacy, and consistency of drug
products. It involves rigorous testing and monitoring at various stages of production to meet regulatory standards and prevent
the release of substandard or harmful medications.
13.Personnel Hygiene
Personal hygieneis crucial in the pharmaceutical industry to prevent product contamination and maintain quality standards. It
involves strict adherence to practices like handwashing, proper attire, and avoidance of certain items in production areas. These
practices help minimize the risk of introducing microbes, particles, and other contaminants that could compromise the safety and
efficacy of medications.
Hygiene Practices:
Handwashing:
Regular and thorough handwashing, especially before entering production areas and after using
the restroom, is essential.
Clothing:
Workers should wear clean, appropriate attire, including gowns or uniforms, to prevent shedding
of skin cells and hair.
Hair and Nail Care:
Hair should be kept short, clean, and restrained, and nails should be trimmed and clean.
Jewelry and Cosmetics:
Jewelry, makeup, and perfume should be avoided in production areas to prevent contamination.
No Food or Drink:
Consumption of food, drinks, or chewing tobacco is strictly prohibited in production, storage, and
laboratory areas.
Health Checks:
Employees should undergo regular health checks to ensure they are free from illness or infections
that could contaminate products.
Importance of Personal Hygiene:
Contamination Prevention:
Personal hygiene practices help prevent the introduction of microorganisms, dust, and other
contaminants that can compromise product quality and safety.
Good Manufacturing Practices (GMP):
Good personnel hygiene is a fundamental aspect of GMP, ensuring that products are
manufactured under controlled conditions.
Product Safety and Efficacy:
By minimizing contamination risks, personal hygiene practices contribute to the safety
and efficacy
Patient Well-being:
Maintaining high standards of personal hygiene ensures that pharmaceutical products are
safe for patient use.
Training and Enforcement:
Training:
Employees should receive thorough training on personal hygiene practices and their
importance.
SOPs:
Standard operating procedures (SOPs) should be implemented and followed by all
personnel to ensure consistency in hygiene practices.
Supervision:
Supervisors and quality assurance personnel should monitor and enforce adherence to
hygiene practices.
Specific Areas of Focus:
Cleanroom Environments:
In cleanrooms, stricter hygiene protocols are necessary to maintain a controlled
environment.
PPE:
Personal protective equipment (PPE), such as gowns, gloves, and hair nets, should be
worn as required in specific areas.
Reporting:
Employees should be encouraged to report any conditions that may adversely affect
product quality.
38.
Productionareas
Weighingareas
Ancillary areas
Storage area
Qualityareas
Equipment's
Materials
Referencestandards etc.has been prescribed.
14.Premises
16.Material
Validated ComputerizedStorage Systems
PM- not to test all batches, but based on
vendor approval and statistical data analysis
Identify test for each container of starting
material (Exception-dedicated facilities)
Reworking of rejected products (new batch
number)
Part of earlier batches into a batch of same
product at defined stages of manufacture
Extension of retesting date
41.
17.Reference Standards
Pharmaceutical manufacturersmust:
•Use official reference standards whenever available.
•Procure Indian Pharmacopoeia reference standards from the Indian Pharmacopoeia Commission.
•Use official reference standards only for the purposes outlined in the relevant monograph.
•Test, release, and store manufacturer-prepared reference standards like official standards.
•Designate a responsible person to oversee the storage of manufacturer-prepared reference standards in
a secure area.
•Establish secondary or working standards through regular testing and checks to maintain
standardization.
To ensure compliance with Good Manufacturing Practices (GMP) for pharmaceutical products,
manufacturers must adhere to the following principles regarding Reference Standards:
IP RS/IS procured from IPC
Procedureforworkingstandard
42.
17.Reference Standards Contd.
Labelreference standards with the following
information:
•Name of the material
•Batch or lot number and control number
•Date of preparation
•Shelf-life
•Potency
•Storage conditions
•Standardize in-house working or secondary standards
against an official reference standard, if available, initially
and periodically.
•Store and use all reference standards to prevent quality
degradation.
43.
18.Waste Material
Properand safe storage of waste materials waiting disposal.
Toxic substances and flammable materials shall be stored in
suitably.
Collect in suitable receptacles for removal to collection points
outside the buildings disposed of safely and in a sanitary
manner at regular and frequent intervals.
The disposal of sewage and effluents (solid, liquid and gas)
shall be in conform the requirements/Pollution Control Board.
All bio-medical waste shall be destroyed as per Bio-Medical
Waste Rules, 2016.
Rodenticides, insecticides, fumigating agents and sanitizing
materials shall be ensured to prevent contamination
45.
Documents shallbe approved, signed and dated by the
responsible persons.
SOPs
Recording entry data shall be clear, spacious, legible
and indelible.
Specifications and testing procedure: Raw material,
Starting and packaging materials, intermediate finished
products.
Master formula records C Batch processing records
Packaging instructions and labels
GDP
19.Documentation
46.
Good practices inproduction are focused on ensuring that operations run
efficiently, with high quality, minimal defects, and in compliance with all
regulations. Key elements include:
•Standard Operating Procedures (SOPs): Establish clear, written SOPs
for every process to ensure consistent and repeatable operations.
•Training: Continuous training of staff on best practices, equipment
operation, safety procedures, and quality standards.
•Preventive Maintenance: Regular maintenance schedules for machinery
and equipment to minimize downtime and maximize operational efficiency.
•Quality Control: Implement frequent in-process quality checks, such as
sampling, testing, and inspections, to ensure that the products meet
predefined specifications.
•Documentation: Ensure that all activities, deviations, and inspections are
properly documented for traceability and regulatory compliance.
20.Good Practices in Production
47.
Establish therequirements for Good practices in QC
Qualification and validation
Control of starting materials, intermediated, and Finished products
Part testing, in case CoA from the reliable manufacturer
Batch record review
Out-of-specification results shall be investigated in accordance with an
approved procedure and record shall be maintained.
Retention Sample-AS/SMF FP, Retest/ expiry + I year.
Other materials minimum 2 years.
GMP-related computerized system shall be validated.
Program for stability studies of finished products to establishing shelf
life.
When required starting materials and intermediate products.
Stability shall be determined prior to marketing and following any
significant changes e.g. changes in in-process, equipment's or
packaging materials.
21.Good Practices in Quality Control
49.
Ensure theadequate qualification for the suitability of
computer hardware and software
Sufficient controls to prevent unauthorized access or
changes to data.
Shall have Controls to prevent omissions in data and
audit trail.
Where critical data are being entered manually, there
shall be an additional check by second person on the
accuracy of the data.
Changes to the computerized system shall be through
change control.
A back-up system shall be ensured to prevent loss of
records
22.Computerized System
50.
Computer System Validation(CSV) is a critical process
in the life sciences industry that ensure computer system
used in the production of pharmaceuticals, medical
devices, and other regulated products are operating
correctly and consistently. CSV is essential for
maintaining the integrity, reliability and security of data
generated by these systems, as well as ensuring
compliance with regulatory requirements.
51.
Pharmacovigilance is avital
component of ensuring drug
safety in the pharmaceutical
industry. It involves the
continuous monitoring of
medication safety throughout
its lifecycle, from
development to post-
marketing surveillance.
Pharmacovigilance (PV) is
responsible for monitoring the
safety of medicines in normal
clinical use and during
clinical trials. Its main aim is
to minimize the risk related to
drugs used and to maximize
their benefits.
Pharmacovigilance