- Multiple sequence alignment (MSA) is the alignment of three or more biological sequences, such as DNA, RNA, or protein sequences. It involves identifying regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences.
- Several algorithms and tools exist for generating MSAs, including ClustalW, T-Coffee, MUSCLE, MAFFT, and MSA. They employ different methods like progressive alignment and iterative refinement. The choice of algorithm depends on factors like number of sequences and computational time.
- MSAs aid in tasks like identifying conserved motifs and domains, phylogenetic analysis, and predicting protein structure and function. Formats like FASTA are used for representing MS







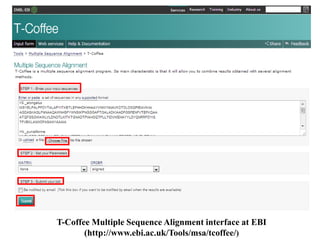






























![1. Go to http://www.ebi.ac.uk/Tools/msa/muscle/ in your
browser.
2. Enter your input sequence.
3. Go to choose file and give your input sequence in any
valid format (GCG, FASTA, EMBL, GenBank, PIR,
NBRF or UniProtKB/Swiss-Prot format).
4. Similarly give other input sequences (There is currently a
limit of 500 sequences and 1MB of data).
5. Click on ‘more options’ button to set the alignment
options. Change output format to clustalW to get results
in aln format. Default value is Pearson/FASTA [fasta].
Output Order -in which the sequences appear in the
final alignment. Default value is: ‘aligned’.
6. Enter submit.](/p?url=https%3A%2F%2Fimage.slidesharecdn.com%2Fmultiplesequencealignment-190706144849%2F85%2FMultiple-sequence-alignment-22-320.jpg&__src=https%3A%2F%2Fwww.slideshare.net%2Fslideshow%2Fmultiple-sequence-alignment-153994579%2F153994579&__type=image)








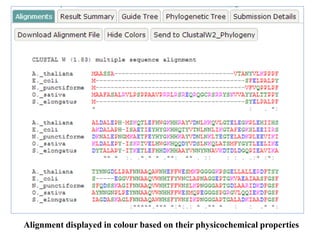
![•Click on ‘more options’ button to set the alignment options
Change the output format ‘clustalw’ (Default value is:
Pearson/FASTA [fasta]).
Matrix
Protein comparison matrix to be used when adding sequences to the
alignment.
Matrix (Protein Only)
Default value is: BLOSUM 62 [bl62]
Gap Open
Penalty for first base/residue in a gap.
Default value is: 1.53
Gap Extension
Penalty for each additional base/residue in a gap.
Default value is: 0.123](/p?url=https%3A%2F%2Fimage.slidesharecdn.com%2Fmultiplesequencealignment-190706144849%2F85%2FMultiple-sequence-alignment-31-320.jpg&__src=https%3A%2F%2Fwww.slideshare.net%2Fslideshow%2Fmultiple-sequence-alignment-153994579%2F153994579&__type=image)
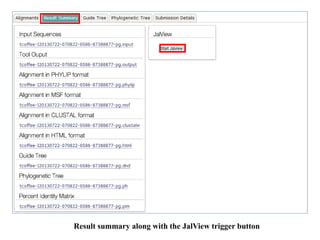
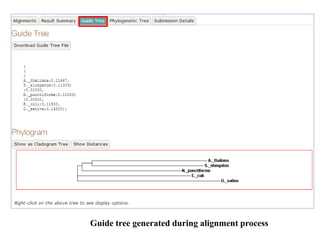
![Order
The order in which the sequences appear in the final alignment
Default value is: aligned
Tree Rebuilding Number
Default value is: 1
Guide Tree Output
Generate guide tree file
Default value is: ON [true]
Max Iterate
Maximum number of iterations to perform when refining the
alignment.
Change the Max Iterate value to ‘2’ to change number of
iterations for better alignment.
Default value is: 0
Perform FFTS (Fast Fourier Transform)
Default value is: local pair
• Click ‘submit’](/p?url=https%3A%2F%2Fimage.slidesharecdn.com%2Fmultiplesequencealignment-190706144849%2F85%2FMultiple-sequence-alignment-32-320.jpg&__src=https%3A%2F%2Fwww.slideshare.net%2Fslideshow%2Fmultiple-sequence-alignment-153994579%2F153994579&__type=image)

































































































